More accurate band gaps
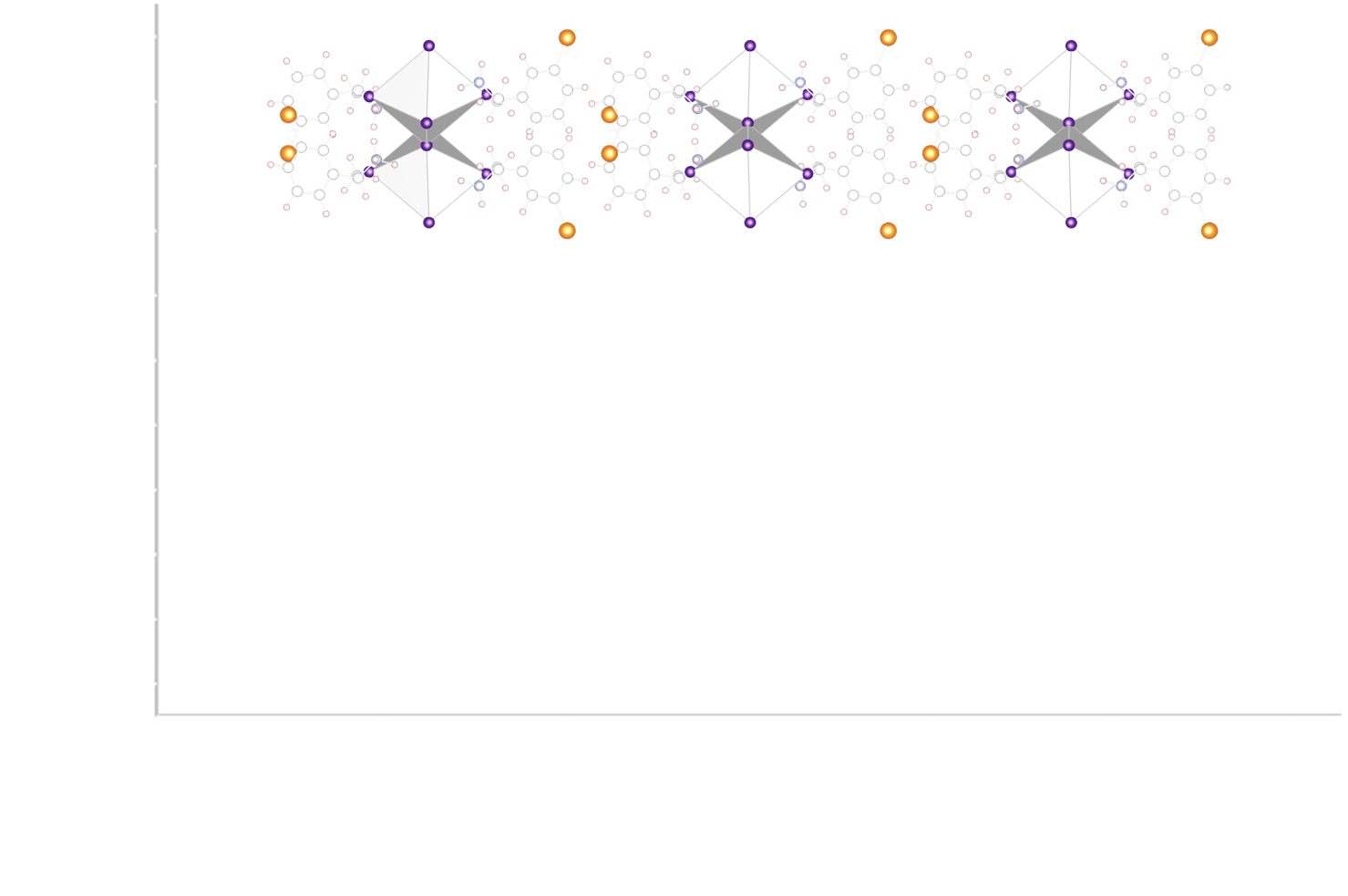
We work mostly with density functional theory (DFT) based approaches for the investigation of semiconductors. It is well known that plain DFT severely underestimates bandgaps and that more accurate values are obtained with either hybrid functionals or GW many-body approaches. However, these approaches are available at a large computational costs and cannot always be used for the large systems we consider such as layered halide perovskites that requires large and sparse simulation cells. For that reason, we are looking for cheapest way to access a more accurate picture of the electronic structure of the materials, such as the so-called DFT-1/2 scheme or the Tran-Blaha modified Becke-Johnson potential.