![]()
Electrochemical Reactivity and Electrosynthesis of Heteroelement Compounds
Electrochemical Reactivity
Electrochemical Methodology
Electrogenerated Reagents
Redox Chemistry of Metallatranes
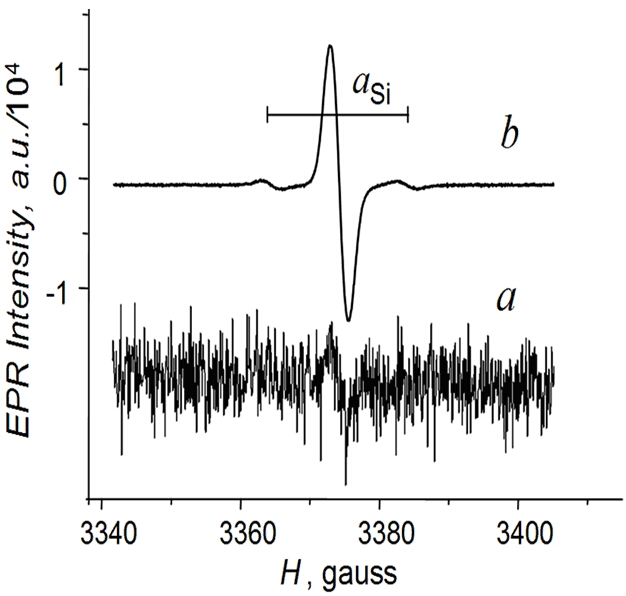
Silyl Radicals
Electrosynthesis
|
|
|
 |
The most surprising result was that these bonds can consecutively give away or accommodate two electrons and this without the cleavage of the M-M linkage! Diode-like behavior of these systems due to mixing the electrons of M=M double bonds with n-electrons of S was demonstrated. |
|
|
||
|
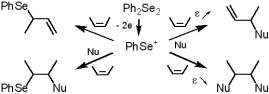
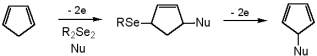
Depending on donor-acceptor interactions through space, easily modulated by polarity of the
reaction media, products of different nature were obtained from the oxidation of
arylalkylselenides. So simple change of proportions of the components of a binary solvent
directs the process to either allylic or a-Nu-substituted products. |
||
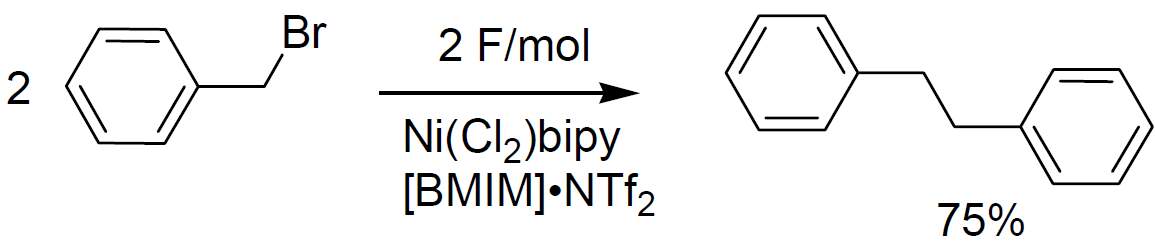
|
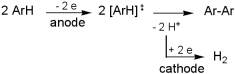
Cathodic Ni(II) cathalysed formation of biaryls and bibenzyls from the corresponding organohalides was shown to be easily and selectively realizable in neat ionic liquids, with no co-solvent added at all. |
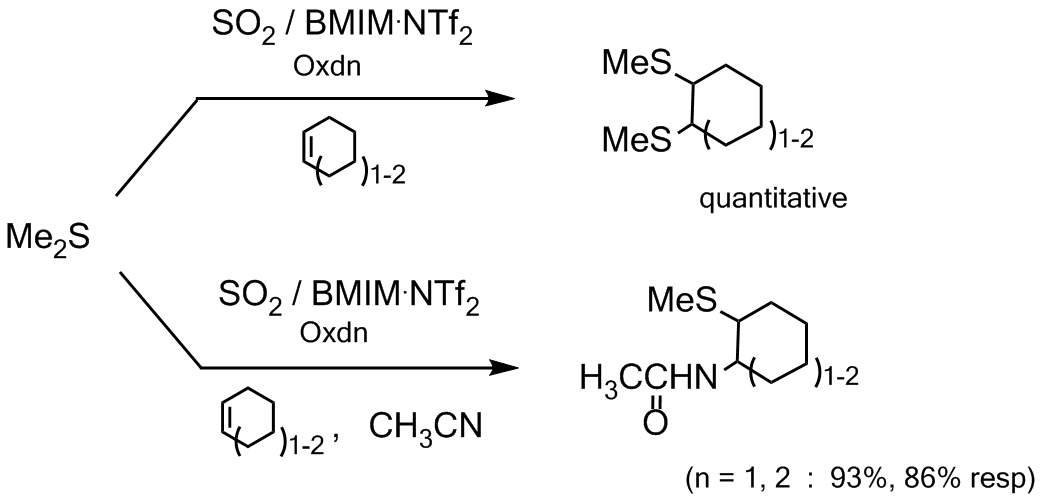 |
Addition of small amount of liquid sulfur dioxide to BMIM NTf2 ionic liquid dramatically decreases the viscosity of this media and allows combining its low nucleophilicity with a good processability and recycling use, the latter is rather easy in this case because the SO2 is a gas at room temperature and all is needed is just to let it evaporate. |
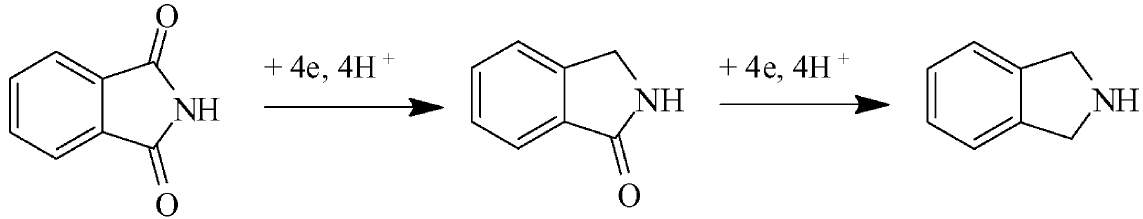 Cathodic decarbonylation of phthalimide is a very convenient way for
preparing isoindoline, the added value of this process is more that 1000
times. The tricky thing here is to master the interplay of hydrogen
evolution and that of the target process. Cathodic decarbonylation of phthalimide is a very convenient way for
preparing isoindoline, the added value of this process is more that 1000
times. The tricky thing here is to master the interplay of hydrogen
evolution and that of the target process.
|
|
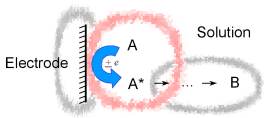
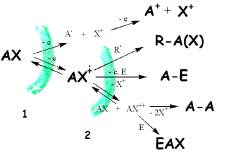 reactions in the bulk of solution..) we
mainly focus on this
primary reactivity, the electrochemical reactivity.
reactions in the bulk of solution..) we
mainly focus on this
primary reactivity, the electrochemical reactivity.
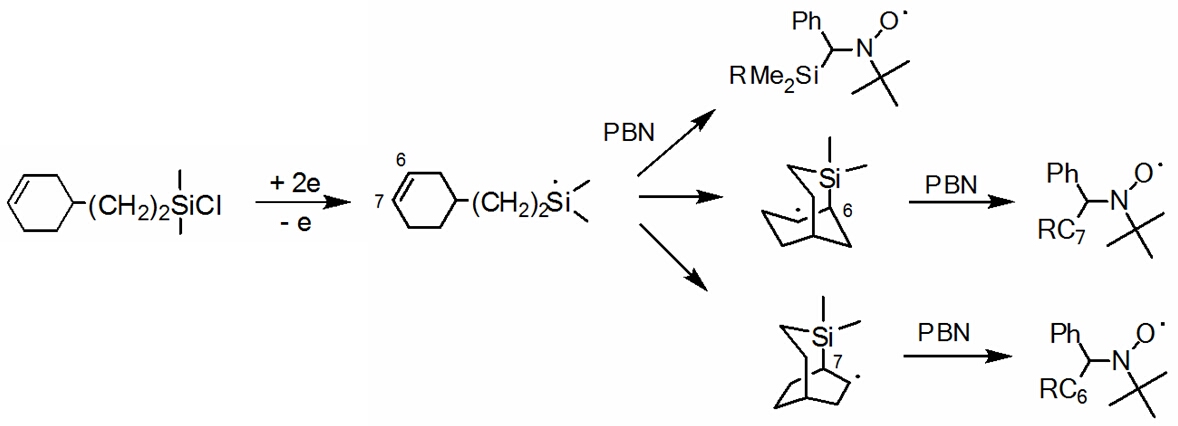
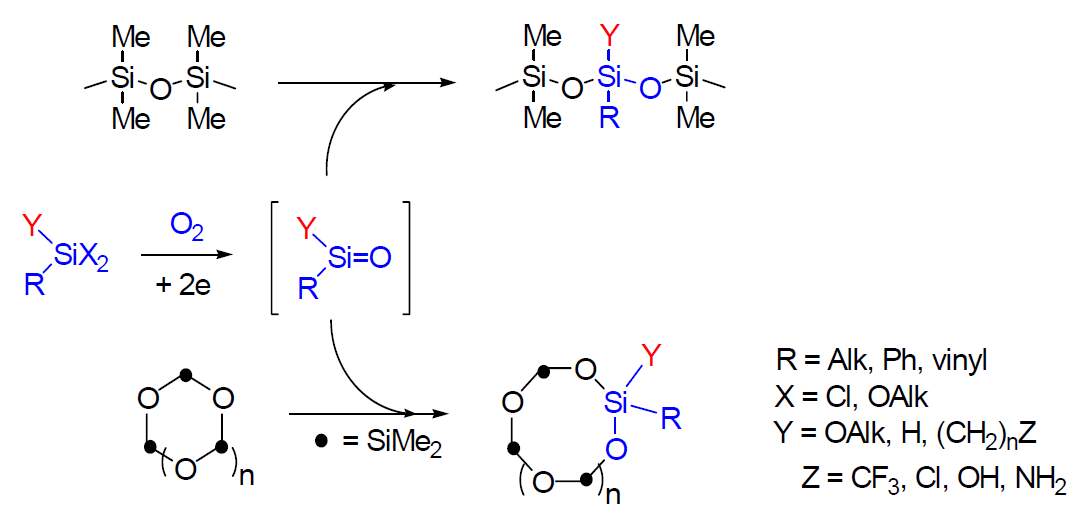
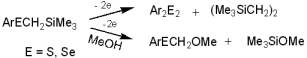
 Organo chlorosilanes can also follow both electron transfer
mechanisms. Here, the stereoelectronic interactions are of
extreme importance: in contrast to carbon-derived systems which form stable p-system
based anion-radicals, specific s-orbital interactions
of the bonds formed by silicon allow anion radicals formation from some totally aliphatic
silicon derivatives.
Organo chlorosilanes can also follow both electron transfer
mechanisms. Here, the stereoelectronic interactions are of
extreme importance: in contrast to carbon-derived systems which form stable p-system
based anion-radicals, specific s-orbital interactions
of the bonds formed by silicon allow anion radicals formation from some totally aliphatic
silicon derivatives. 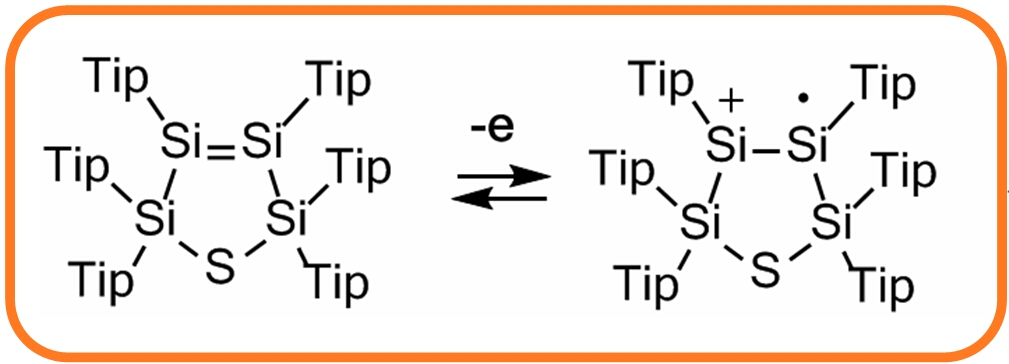
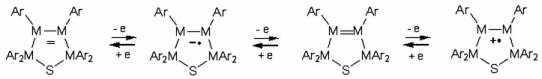


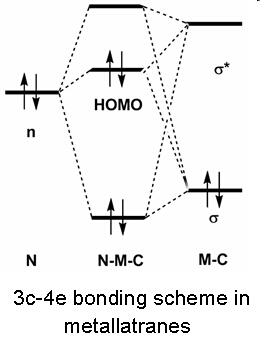
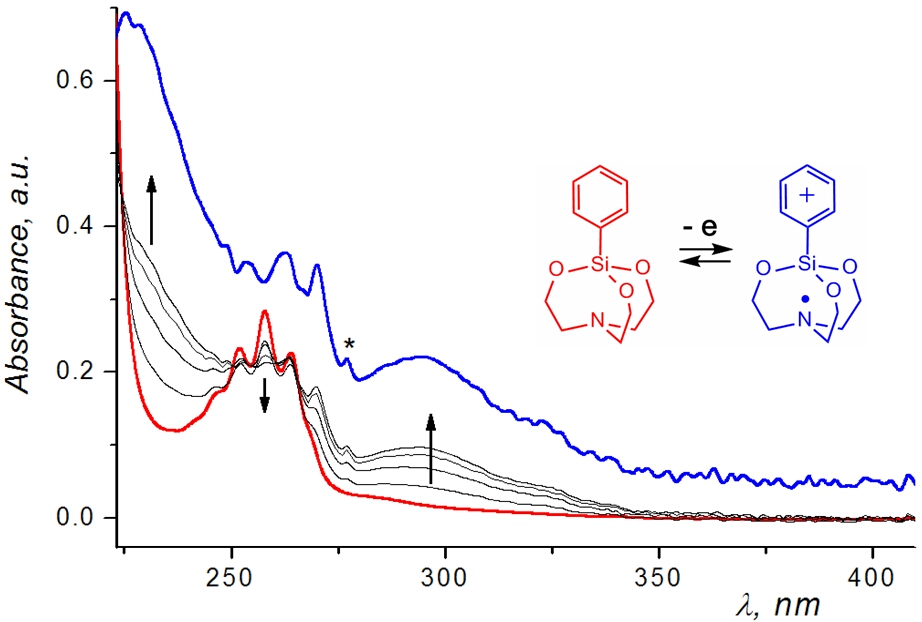
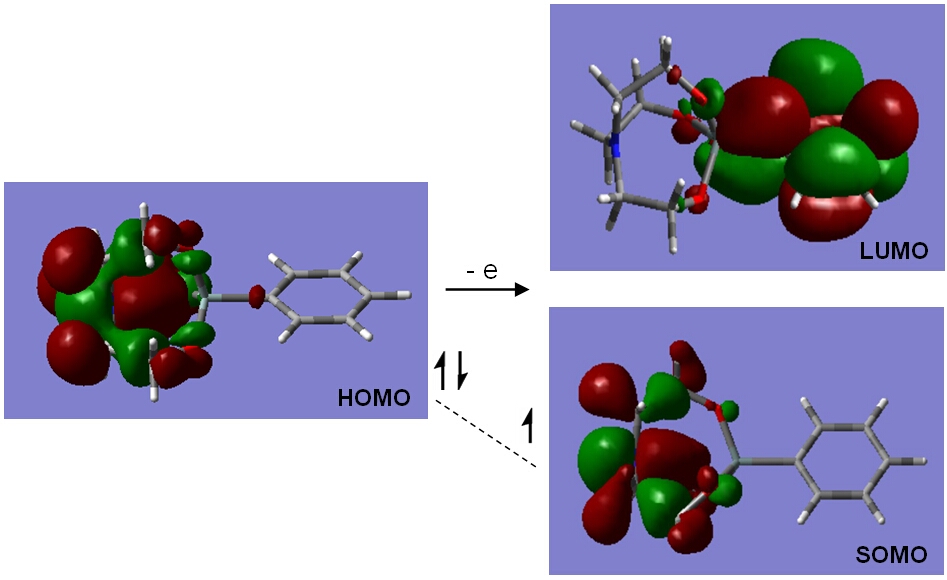
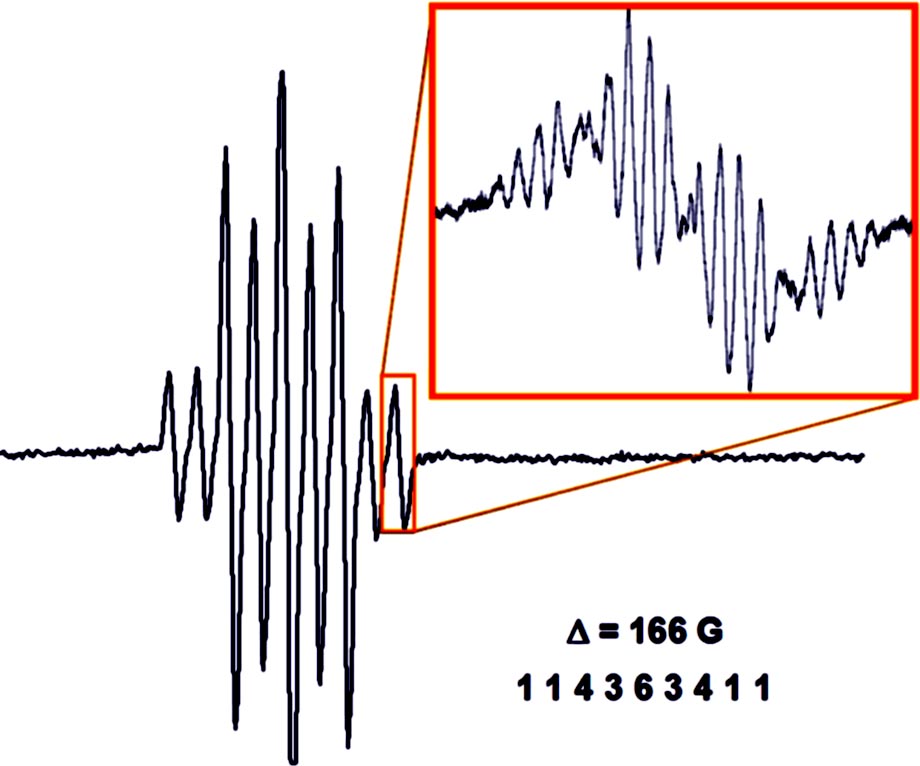
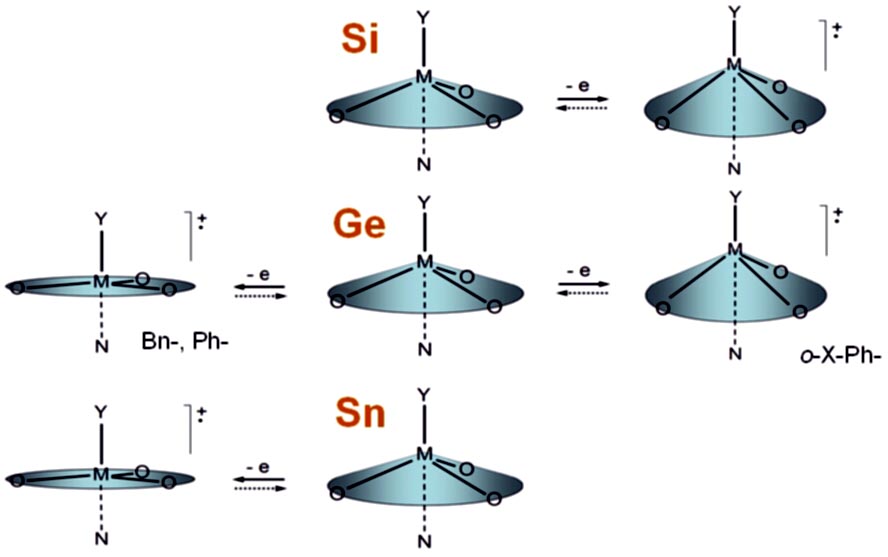
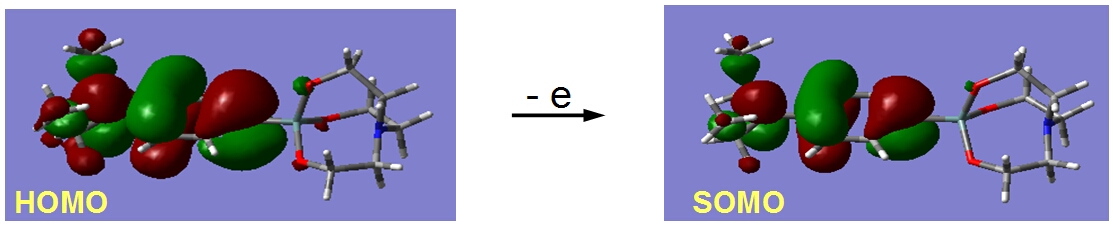
 In contrast to aryl- and vinyl-substituted metallatranes, hyperconjugation of
In contrast to aryl- and vinyl-substituted metallatranes, hyperconjugation of 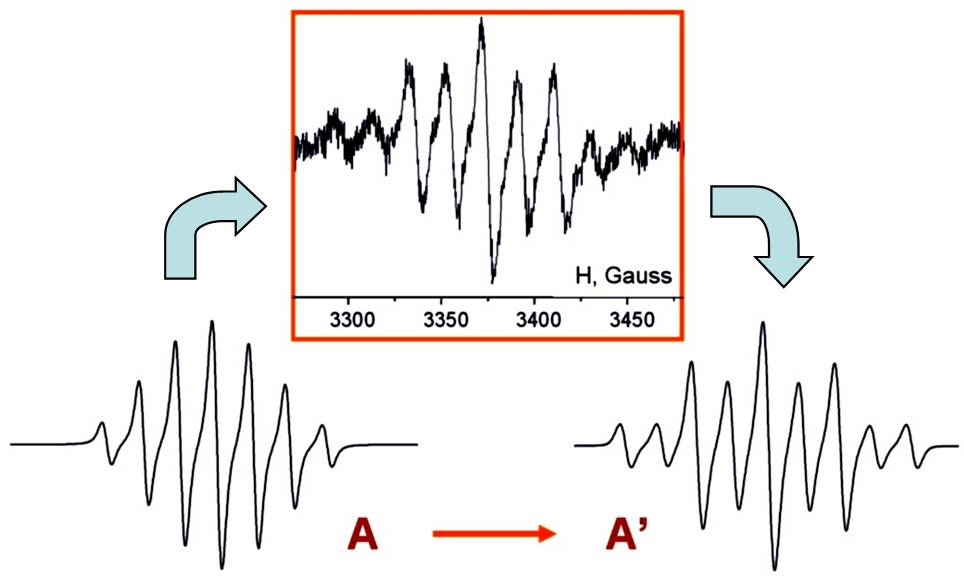 ESR-spectroelectrochemistry also enabled us to make evident the process of penta-
to hexa-coordination changes in the cation radicals of arylgermatranes.
Primary cation radicals of o-Br and o-F-Ph germatranes are formed in the
geometry close to that of the starting molecule and then slowly evolve to
adopt 6-coordinated configuration with additional coordination from ortho-halogen
substituent. Kinetics and activation parameters of this process were measured
as well.
ESR-spectroelectrochemistry also enabled us to make evident the process of penta-
to hexa-coordination changes in the cation radicals of arylgermatranes.
Primary cation radicals of o-Br and o-F-Ph germatranes are formed in the
geometry close to that of the starting molecule and then slowly evolve to
adopt 6-coordinated configuration with additional coordination from ortho-halogen
substituent. Kinetics and activation parameters of this process were measured
as well.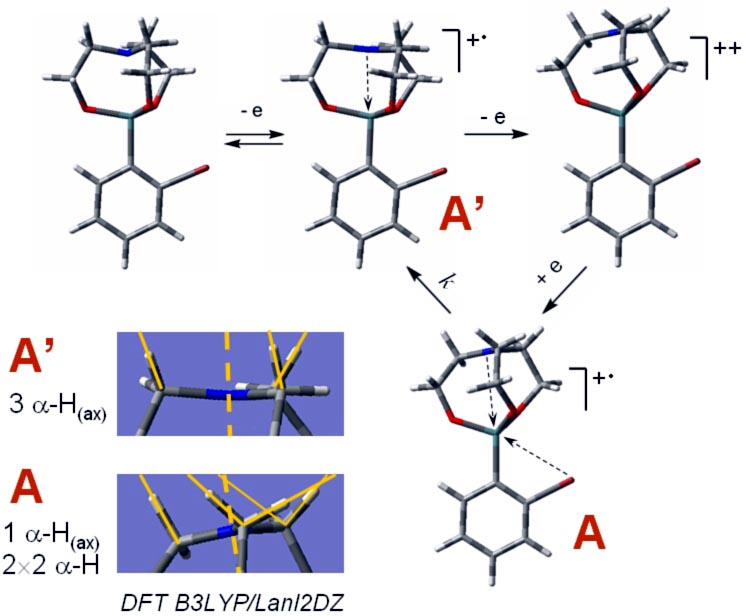

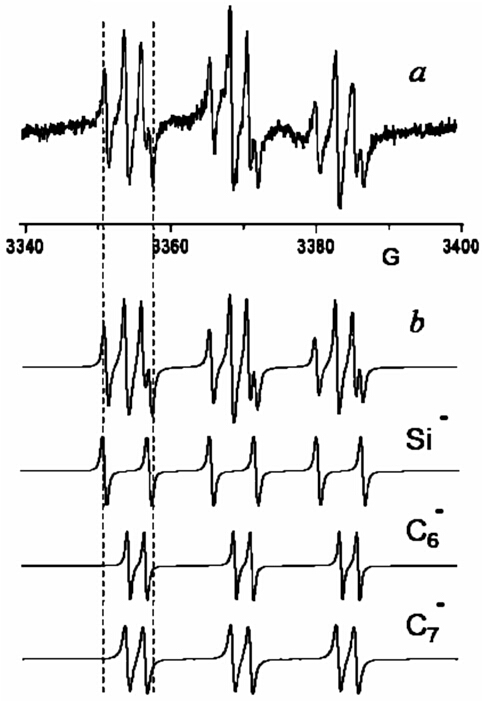
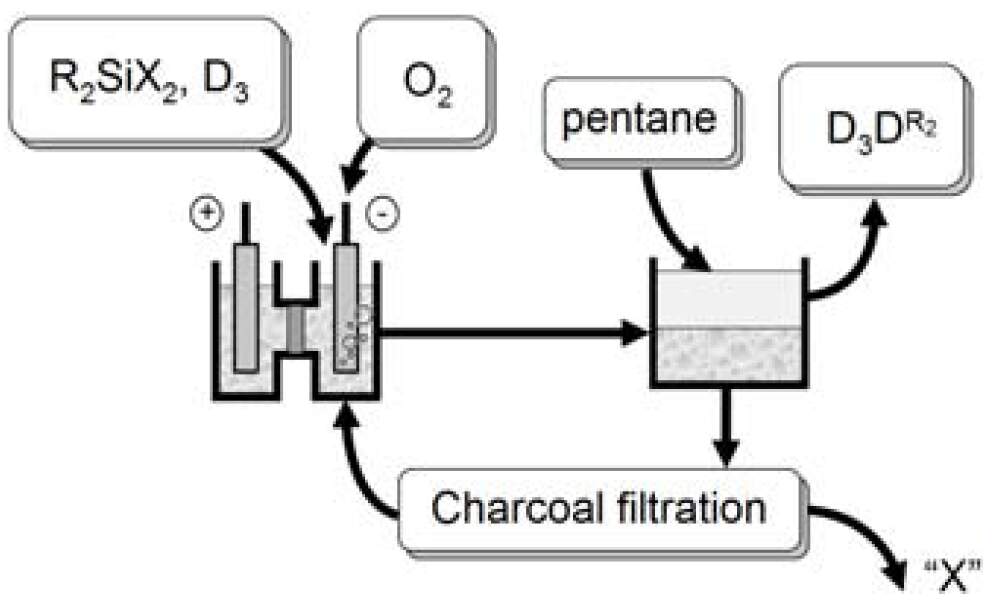
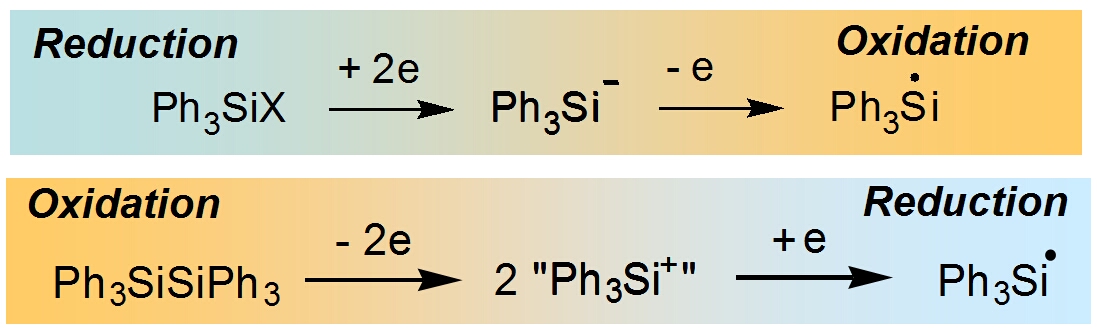 To study these apparently 2-electron systems, we used the principle of
"reversed electrochemistry" in a two-compartment flow-through
electrochemical cell – when first realizing the overall 2e process in one
part of this cell and then making one step back in the second compartment by
applying the potential of the opposite polarity. Now, same silyl radicals R3Si
can be prepared staring from both sides – either oxidation or reduction of
neutral organosilicon molecules, provided that they are electrochemically
active.
To study these apparently 2-electron systems, we used the principle of
"reversed electrochemistry" in a two-compartment flow-through
electrochemical cell – when first realizing the overall 2e process in one
part of this cell and then making one step back in the second compartment by
applying the potential of the opposite polarity. Now, same silyl radicals R3Si
can be prepared staring from both sides – either oxidation or reduction of
neutral organosilicon molecules, provided that they are electrochemically
active.